Enfermedades genéticas
definición
Una enfermedad genética o hereditaria es una enfermedad causada por uno o más genes de la persona en cuestión. El ADN actúa aquí como un desencadenante directo de la enfermedad. Para la mayoría de las enfermedades genéticas, se conocen las ubicaciones de los genes causantes. Si se sospecha una enfermedad genética, el diagnóstico correspondiente se puede realizar mediante un examen genético.
Por otro lado, también hay una serie de enfermedades cuya aparición tiene influencia genética o se discute, como la diabetes mellitus (“diabetes”), la osteoporosis o la depresión. Estas son las llamadas disposiciones, es decir, una mayor probabilidad de ciertas enfermedades. Las disposiciones deben distinguirse de las enfermedades hereditarias.

Estas son enfermedades hereditarias comunes.
En términos absolutos, las enfermedades hereditarias no son comunes, pero las enfermedades hereditarias enumeradas aquí ocurren con frecuencia en comparación con otras enfermedades de causa genética.
-
El síndrome de Marfan
-
Anemia falciforme
-
Hemofilia (hemofilia A o B)
-
Mutación del factor V Leiden y resistencia a APC resultante
-
Rojo verde debilidad
-
Deficiencia de glucosa-6-fosfato deshidrogenasa (deficiencia de G6PD)
-
Polidactilia ("dedos múltiples", también posible como síntoma en otras enfermedades)
-
Trisomía 21 (síndrome de Down)
-
Chorea Huntington
causas
Las enfermedades hereditarias son extremadamente diversas en su apariencia. Básicamente, solo tienen una cosa en común: la causa de cada uno de ellos reside en el ADN, es decir, en el material genético de la persona en cuestión. Aquí pueden ocurrir varios cambios, como mutaciones (intercambio de información de ADN) o deleciones (falta de cierto material genético).
Una gran cantidad de información está codificada en el material genético, como los "planos" de varios componentes que son importantes para el funcionamiento de una célula del cuerpo. Estos pueden ser enzimas, canales de electrolitos o sustancias mensajeras, por ejemplo. Estos elementos más pequeños se leen entonces incorrectamente o no se leen del ADN, que luego falta en el sofisticado sistema del cuerpo. La información genética incorrecta o faltante, por lo tanto, causa ciertas disfunciones en el cuerpo. Estos luego causan síntomas según el sistema funcional en el que ahora falta un elemento.
Descubra todo sobre el tema aquí: La prueba genética.
Así se heredan las enfermedades hereditarias
Todas las enfermedades hereditarias se heredan de forma monogenética o poligénica: esto significa que hay una o más ubicaciones genéticas que deben cambiarse para provocar una enfermedad.
Además, los rasgos genéticos siempre pueden heredarse de manera dominante o recesiva: recesivo significa que debe haber una predisposición para esta enfermedad hereditaria particular tanto en los genes paternos como maternos. En el caso de la herencia dominante, un cambio (es decir, uno de los padres) es suficiente para desencadenar la enfermedad. De ello se deduce que con las enfermedades de herencia dominante, las personas que son portadoras también se enfermarán, mientras que con una herencia recesiva, por lo general, ni siquiera se sabe que existe una predisposición genética correspondiente.
También existen enfermedades que se heredan a través de los cromosomas sexuales, como la hemofilia o la ceguera rojo-verde. Las facilidades para esto generalmente se encuentran en el cromosoma X, ya que el cromosoma Y es muy pequeño en general y generalmente puede almacenar poca información genética. Por tanto, se habla de enfermedades hereditarias ligadas al cromosoma X. Por lo general, estos afectan significativamente más a los hombres que a las mujeres, ya que las mujeres pueden compensar cualquier información incorrecta sobre el cromosoma X con el segundo.
La forma exacta en que se hereda una enfermedad genética suele ser fácil de investigar si está interesado.
Pruebas antes del nacimiento
En principio, el material genético del niño ya puede examinarse en el útero para todas las enfermedades hereditarias cuyas ubicaciones genéticas causales se conocen. Sin embargo, los análisis genéticos requieren mucho tiempo, por lo que generalmente solo se analiza la ubicación sospechosa del gen; para esto, a su vez, debe haber una sospecha justificada de una enfermedad genética.
Para tal examen, se puede extraer material genético del líquido amniótico o de la placenta y utilizarlo para el análisis.
Sin embargo, siempre hay que tener en cuenta que cualquier diagnóstico invasivo también implica un riesgo para la vida del feto. Por tanto, dichos pinchazos deben pesarse individualmente en cada caso.
También hay medidas que pueden indicar una enfermedad genética, como la medida de la transparencia de la nuca como signo de trisomía 21. Estos métodos no son peligrosos para el feto, pero no pueden ofrecer una certeza absoluta sobre la presencia de una enfermedad genética. Por tanto, aquí también se debe considerar cuidadosamente una operación.
Trisomía 21
La causa de la trisomía 21 es el cromosoma 21, que no está presente dos veces sino tres veces en las personas afectadas. Esta variante del ADN se crea cuando los cromosomas se distribuyen en las células germinales parentales, es decir, los espermatozoides o los óvulos. Por tanto, es un "error de distribución" y no un cambio en el material genético real. Esto explica por qué la trisomía 21 puede ocurrir de manera espontánea en todas las familias y por qué la probabilidad de tener un hijo con síndrome de Down es la misma en todas las familias. Estrictamente hablando, la trisomía 21, como otras trisomías, no debe contarse como una enfermedad hereditaria en el verdadero sentido. Sin embargo, la trisomía 21 es la enfermedad relacionada con el ADN más común en los recién nacidos.
Las características del conjunto modificado de cromosomas en el síndrome de Down ya se pueden ver en el feto en el útero: los retrasos en el crecimiento y los defectos pueden provocar, entre otras cosas, un cráneo demasiado pequeño, huesos cortos del muslo y la parte superior del brazo y defectos cardíacos. Una gran cantidad de líquido amniótico también puede ser una indicación de trisomía 21, ya que los niños no nacidos afectados beben o tragan relativamente poco líquido amniótico. Sin embargo, ¡ninguna de estas características son signos definitivos del síndrome de Down!
Además de los signos de retraso del crecimiento mencionados, los niños con síndrome de Down a menudo también muestran un retraso en el desarrollo, por ejemplo, en las áreas del lenguaje y las habilidades motoras. Las personas afectadas por el síndrome de Down a menudo muestran habilidades sociales notables, mientras que la inteligencia a menudo se mantiene por debajo del promedio. Sin embargo, las personas afectadas difieren mucho en estas características y no es raro que se gradúen de la escuela después de recibir un buen apoyo.
Más adelante en la vida, las personas con trisomía 21 tienen un mayor riesgo de ser diagnosticadas con ciertas enfermedades. Estos incluyen la enfermedad de Alzheimer, la epilepsia y el cáncer, particularmente la leucemia. No obstante, la esperanza de vida de las personas con síndrome de Down sigue aumentando: mientras tanto, las personas afectadas suelen llegar a los 60 o 70 años.
Puede encontrar más información en nuestro sitio web Síndrome de Down
Deficiencia de alfa-1 antitripsina
La deficiencia de alfa-1 antitripsina puede tomar diferentes formas y formas, dependiendo de las características genéticas exactas de la persona afectada. Esto significa que no todas las deficiencias de alfa-1 antitripsina producen síntomas. A continuación, solo se discutirá el tipo clínicamente conspicuo (PiZZ) de esta enfermedad determinada genéticamente.
El defecto enzimático presente en esta enfermedad provoca la degradación y remodelación de los componentes básicos del tejido orgánico de las personas afectadas. Además, el hígado filtra las proteínas defectuosas de la sangre y se acumulan allí. Esto puede provocar inflamación del hígado (hepatitis), cirrosis o cáncer de hígado. Las vías respiratorias de los pulmones se vuelven inestables debido a la falta de tejido estable y colapsan más rápidamente: se desarrolla el cuadro clínico de la EPOC (enfermedad pulmonar obstructiva crónica). Este cuadro clínico suele ser el primer síntoma de la deficiencia de alfa-1 antitripsina, por lo que cualquier persona con EPOC a una edad más temprana debe ser examinada para detectar una deficiencia de alfa-1 antitripsina.
Si la enfermedad ha persistido durante mucho tiempo, los pulmones pueden sobreinflarse, ya que el aire que respira no se puede exhalar correctamente a través de las vías respiratorias inestables y se acumula en los pulmones. Como terapia, además de evitar constantemente fumar cigarrillos y vacunas regulares para prevenir enfermedades respiratorias, también se deben tomar medidas medicinales: La alfa-1-antitripsina faltante puede administrarse por vía intravenosa para aliviar los síntomas en la medida de lo posible y detener el curso de la enfermedad.
Puede encontrar más información en nuestro sitio web Deficiencia de alfa-1 antitripsina
hemofilia
El grupo de hemofilia también se conoce coloquialmente como “hemofilia”, ya que este término describe con mucha precisión el síntoma principal de esta enfermedad hereditaria: las personas afectadas sangran más tiempo y, dependiendo de la gravedad de la enfermedad, más a menudo que no afectadas.
El sangrado generalmente se detiene mediante lo que se conoce como cascada de coagulación, una vía de señalización endógena que evita la pérdida excesiva de sangre. En este sistema de coagulación intervienen 13 factores, que se activan uno tras otro. Esto se puede imaginar como una serie de fichas de dominó: si golpeas una piedra (factor de coagulación), activa la siguiente, y así sucesivamente. Al final de esta ruta de señal o del dominó hay coagulación sanguínea. Con la hemofilia, falta un factor determinado, según el subtipo específico de la enfermedad: la reacción en cadena se interrumpe aquí.
La terapia para la enfermedad se puede llevar a cabo determinando el factor faltante y agregándolo desde el exterior. Por lo tanto, las personas afectadas deben inyectarse regularmente un preparado con este factor de coagulación para que pueda tener lugar el resto de la reacción en cadena.
Puede encontrar más información en nuestro sitio web Enfermedad de la sangre
Fibrosis quística
En la enfermedad genética fibrosis quística, también conocida como fibrosis quística, hay una producción defectuosa de canales iónicos, más precisamente de canales de cloruro. Como resultado, la composición de las secreciones corporales (por ejemplo, sudor, secreciones del tracto respiratorio y el páncreas) de las personas afectadas cambia: dado que la falta de cloruro significa que se introduce menos agua en el conducto de la glándula respectiva, la secreción es relativamente viscosa.
Como resultado, los síntomas generalmente se desarrollan en el tracto digestivo, ya que la secreción con las enzimas digestivas no puede fluir bien desde el páncreas al intestino y, por lo tanto, daña el páncreas mismo. Además, son comunes los trastornos digestivos como heces grasas, diarrea y el bajo peso corporal resultante.
El segundo gran grupo de síntomas generalmente se desarrolla en los pulmones: dado que el moco que se produce naturalmente en los pulmones es más viscoso que en las personas sanas, es más difícil eliminarlo de los cilios. Esto puede provocar tos crónica y obstrucciones de los bronquios (bronquiectasias). La mayor cantidad de secreción pulmonar también proporciona un buen ambiente para el crecimiento de bacterias, lo que resulta en infecciones respiratorias frecuentes y neumonía.
La fibrosis quística se trata sintomáticamente con expectorantes, enzimas digestivas y antibióticos para infecciones.
Puede encontrar más sobre esto en nuestro sitio web. Fibrosis quística
Factor V Leiden y resistencia a APC
Una mutación del factor V Leiden implica un cambio en la información genética que puede causar un aumento de la coagulación sanguínea. La razón de esto es el factor V en la llamada cascada de coagulación del cuerpo: esta ruta de señal asegura que en caso de una lesión, la herida sea cerrada por las propias "proteínas adhesivas" del cuerpo (fibrina). Hay 13 factores en esta ruta de señalización, que se nombran con números romanos (¡significa “sufrimiento del factor 5”!). El factor V tiene un efecto beneficioso sobre la formación de un tapón de fibrina, pero también puede ser inhibido por la llamada proteína C activada (APC para abreviar). Esto juega un papel importante en la regulación de esta vía de señalización y en la prevención de la coagulación sanguínea excesiva.
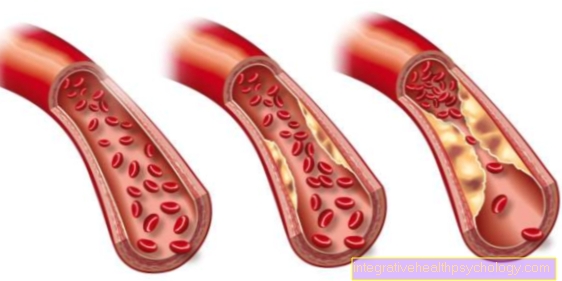
El factor V mutado está presente en los individuos afectados pero no responde a la APC. El cuerpo carece de un "dispositivo de seguridad" importante en este punto para evitar la coagulación de la sangre sin motivo, que puede incluso bloquear los vasos y, por lo tanto, causar trastornos circulatorios.
En términos estadísticos, las personas que se ven afectadas por una mutación del factor V de Leiden tienen más probabilidades de experimentar un evento trombótico (es decir, una trombosis o embolia pulmonar), incluso sin antecedentes de factores de riesgo típicos. En términos técnicos, también se habla de “trombofilia”, es decir, tendencia a la coagulación.
Puede encontrar más sobre esto en nuestro sitio web. Factor V Leiden
Enfermedad de Gaucher
En la enfermedad de Gaucher, el cambio en la información del ADN provoca un defecto en una enzima involucrada en el metabolismo de los lípidos, más precisamente la glucocerebrosidasa: esto ayuda a descomponer los componentes de las células viejas. En caso de un defecto, puede haber una reducción de la funcionalidad o incluso una pérdida de funcionalidad y, en consecuencia, los síntomas aparecen en la infancia o la edad adulta.
Los síntomas de la enfermedad de Gaucher se deben en gran parte a un agrandamiento del hígado y el bazo, cuyo crecimiento el cuerpo intenta compensar la falta de enzimas. Esto aumenta la degradación de todos los componentes sanguíneos, que pueden reconocerse en el hemograma y utilizarse como indicador de diagnóstico junto con el hígado y el bazo agrandados.
La enzima glucocerebrosidasa faltante puede usarse terapéuticamente como fármaco. El pronóstico y el curso de la enfermedad de Gaucher dependen en gran medida de la gravedad de la pérdida de función de la enzima.
Para obtener más información, siga leyendo aquí.: Enfermedad de Gaucher.
Enfermedad de osler
La enfermedad de Osler es una enfermedad hereditaria que se caracteriza por una fuerte vasodilatación. En principio, esta expansión de los vasos puede ocurrir en cualquier lugar, tanto en la piel como en los órganos internos. Las paredes de los vasos agrandados son relativamente delgadas y se desgarran fácilmente. Como resultado, las áreas afectadas sangran rápidamente.
La vasodilatación se presenta con especial frecuencia en la cara y en la mucosa nasal, por lo que las personas afectadas suelen quejarse de hemorragias nasales frecuentes y pequeñas hemorragias con manchas en la cara.
Si se sospecha la enfermedad de Osler, se deben realizar los diagnósticos adecuados, ya que la vasodilatación también puede ocurrir en órganos vitales u órganos con buen riego sanguíneo, como los pulmones, el cerebro o el hígado, en los que el sangrado de un vaso roto es peligroso.
Puede encontrar más sobre este tema en nuestro sitio web. Enfermedad de osler
Enfermedad de Recklinghausen
La neurofibromatosis tipo 1, o enfermedad de Recklinghausen, es una enfermedad genética en la que los afectados suelen desarrollar tumores en las células de la cubierta nerviosa. Los tumores que se desarrollan pueden ser tanto benignos como malignos y aparecen a una edad temprana.
Sin embargo, los tumores típicos son los neurofibromas benignos: consisten en células que envuelven y aíslan el nervio como un cable eléctrico, así como el tejido conectivo circundante. Son benignos, es decir, tumores que no se propagan y crecen lentamente.
Sin embargo, la cirugía para extirpar neurofibromas puede ser difícil, ya que a menudo están firmemente adheridos al nervio y luego se debe extirpar el nervio correspondiente. Sin embargo, esta es la única opción de tratamiento para el neurofibroma sintomático, ya que no es posible la terapia causal para esta enfermedad hereditaria.
Puede encontrar más sobre este tema en nuestro sitio web. Neurofibromatosis tipo 1
Distrofia muscular
El término distrofia muscular describe un grupo de enfermedades hereditarias en las que ciertos componentes musculares no pueden o no pueden ser ensamblados correctamente por las células del cuerpo. Como resultado, las personas afectadas suelen desarrollar debilidad muscular desde la niñez y la adolescencia, y esto puede resultar en una pérdida de masa muscular, restricciones de movimiento e incluso discapacidades físicas.
Si se sospecha la presencia de distrofia muscular, primero se deben determinar los valores sanguíneos. Si los valores coinciden con el diagnóstico sospechado, aún se puede realizar una biopsia de músculo: se toma una pequeña muestra de tejido del músculo, que luego se examina microscópicamente en busca de defectos celulares. También es posible realizar un examen genético para establecer el diagnóstico, ya que las localizaciones genéticas correspondientes suelen ser conocidas para las diversas formas de distrofia muscular y tendrían que cambiarse. No se conoce una terapia causal para las distrofias musculares.
Puede encontrar más sobre este tema en nuestro sitio web. Distrofia muscular
Xeroderma pigmentoso
Xeroderma pigmentosum es una rara enfermedad hereditaria en la que ciertas enzimas de la piel de la persona afectada no funcionan. Estas enzimas normalmente se encargan de la reparación en el ADN, que puede resultar dañado por la luz solar o la luz UVB contenida. El daño de los rayos UVB puede causar cáncer de piel en las personas afectadas, así como en todas las demás personas, pero con Xeroderma Pigmentosum el proceso se acelera por la falta de mecanismos de reparación. Como resultado, las personas afectadas desarrollan formas graves de cáncer de piel en la infancia y la adolescencia y después de una breve exposición a la luz solar.
Aún no es posible una terapia causal. Las personas afectadas tienen que evitar la luz solar de por vida, por lo que el apodo de "niños claros de luna" se ha establecido para las personas afectadas (a veces muy jóvenes) afectadas. Además, estas personas deben ser supervisadas por un dermatólogo para la detección periódica del cáncer de piel con el fin de eliminar el cáncer de piel recientemente desarrollado de inmediato. Si se siguen estrictamente estas medidas, la esperanza de vida de una persona con xeroderma pigmentoso es aproximadamente la misma que la de una persona no afectada.
Puede encontrar más sobre esta enfermedad en nuestro sitio web. Xeroderma pigmentoso
Síndrome de Lynch
El síndrome de Lynch es un cambio en el ADN que causa una enzima defectuosa en las células del cuerpo.En las personas afectadas, por lo tanto, un cierto mecanismo es defectuoso, que de otro modo se supone que protege a las células de la degeneración, es decir, del crecimiento descontrolado; las personas con síndrome de Lynch, por lo tanto, tienen un riesgo mucho mayor de desarrollar cáncer.
El cáncer de colon a menudo ocurre porque las células a menudo se dividen naturalmente aquí de todos modos y los errores en la programación de crecimiento y muerte de una célula se hacen evidentes más rápidamente. Las personas afectadas a menudo desarrollan un tumor en el intestino grueso a una edad inusualmente joven, es decir, antes de los 50 años, que luego se denomina HNPCC (cáncer de colon hereditario no pólipo). Sin embargo, no todas las personas que tienen la estructura genética del síndrome de Lynch desarrollarán cáncer de colon. Por otro lado, otros órganos también pueden desarrollar un tumor, ya que las predisposiciones genéticas que favorecen el desarrollo de un tumor están presentes en todas las células del cuerpo. Por lo tanto, las personas afectadas por el síndrome de Lynch necesitan controles periódicos y exámenes preventivos para tratar adecuadamente los tumores que se desarrollan en una etapa temprana.
Puede encontrar más sobre este tema en nuestro sitio web. Síndrome de Lynch









.jpg)